
Recent Pathological Diagnoses of Several Rare Pediatric Tumors or Diseases
In recent years, some rare tumors or diseases that predominantly affect children and adolescents have been successively reported in the literature. Most of these diseases have unique clinicopathological and molecular biological characteristics, and their treatment plans and prognoses also vary. Therefore, recognizing and accurately diagnosing these diseases is particularly important. This article will introduce several rare tumors or diseases in children encountered in daily pathological diagnostic work, in order to help pathologists, especially pediatric pathologists, further understand the pathological features of these diseases and provide a basis for pathological diagnosis and research.
Undifferentiated round cell sarcoma with CIC-DUX4 translocation
A tumor of the peripheral primitive neuroectodermal tumor group, commonly seen in children and young adults, with a predilection for the limbs; other sites such as the trunk, head and neck, brain, and pelvis have also been reported. Clinically presents as a rapidly growing, superficial or deep soft tissue mass, with a poor prognosis; most patients die within 2 years, and the tumor has a high metastasis rate.
Histological features include sheets of densely packed small round tumor cells, moderate nuclear pleomorphism, coarse chromatin, prominent nucleoli, indistinct cytoplasmic borders. Focal clear cytoplasm can be seen in tumor cells, mitotic figures are easily found, sometimes spindle-shaped tumor cells and geographic necrosis are observed, locally may appear myxoid or arranged in cords. Immunohistochemical staining is nonspecific: positive for CD99, WT1, Fli-1, ERG, INI1, etc.; negative for S-100 protein, cytokeratin (CK), desmin, myogenin, TLE1, etc. Molecular genetic examination reveals chromosomal t(4;19)(q35;q13.1) translocation in most cases; other minority cases may have t(10;q26;q13) translocation. Fluorescence in situ hybridization (FISH) examination may show CIC-DUX4 translocation (Figure 1,Figure 2)。
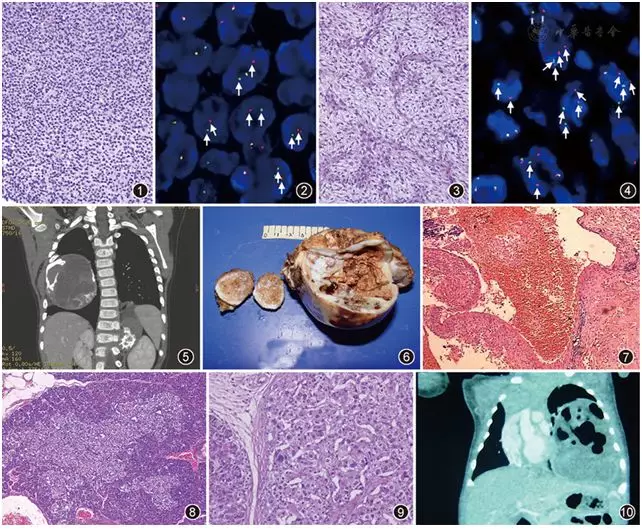
▲Figure 1Small round cell undifferentiated tumor, tumor cells arranged in sheets. HE, low magnification
▲Figure 2FISH detection of CIC gene status (dual-color break-apart probe). Arrows in the image indicate positive CIC gene break, showing separated red and green signal dots in the cells. FISH, ×1,000
▲Figure 3Spindle tumor cells with clear cytoplasm, arranged in solid sheets, with abundant, delicate, curved vascular structures in between. HE, low magnification
▲Figure 4FISH detection of FUS gene status (dual-color break-apart probe). Arrows in the image indicate positive FUS gene break translocation, showing separated red and green signal dots in the cells. FISH, ×1,000
▲Figure 5CT scan shows a chest wall mass occupying most of the right thoracic cavity, with a few high-density bone and cartilage tissues inside.
▲Figure 6Gross photograph shows a cystic-solid mass with cartilage and bone tissue.
▲Figure 7Vascular-like cystic cavities, cyst walls composed of fibrous tissue. HE, low magnification
▲Figure 8Large islet structure with relatively clear boundaries. HE, low magnification
▲Figure 9Shows partial islet cell nuclei enlarged and hyperchromatic. HE, medium magnification
▲Figure 10Gross photograph shows a cystic-solid mass, partially spongy, with a fibrous capsule and clear demarcation from surrounding lung tissue.
Low grade fibromyxoid sarcoma
A low-grade malignant fibroblastic tumor composed of spindle cells, collagen, or myxoid matrix, with specific FUS-CREB3L2 or FUS-CREB3L1 fusion gene abnormalities in most cases. Commonly occurs in young adults, 20% of cases are under 18 years old, frequently seen in limbs and trunk, occasionally in visceral organs. Clinically presents as a painless, slowly growing mass, often for over 1 year; masses are mostly located in deep soft tissues, while in children they often occur in superficial soft tissues. Tumor progression is slow, with low local recurrence and metastasis rates within a few years after surgery, but recurrence and metastasis rates increase with longer follow-up. Common metastatic sites include lung, pleura, and chest wall. Superficial masses have a better prognosis.
Microscopic examination shows a well-defined mass, often with tumor infiltration into surrounding soft tissues. Tumor cells are spindle-shaped with indistinct cytoplasm, nuclei slightly enlarged or hyperchromatic, mitotic figures are rare. Fibrous and myxoid areas are mixed. The fibrous area is hypocellular, rich in delicate fibers or collagen matrix, showing short fascicles and whorled structures; sometimes rosette-like structures surrounded by hypocellular collagen are seen. The myxoid area is more cellular, with more prominent blood vessels; perivascular areas may be hypercellular, with varying degrees of perivascular sclerosis around small vessels. Necrosis is rare; cystic changes, osseous metaplasia, and staghorn vessels may be seen. Sometimes focal hypercellular areas, mitotic figures, pleomorphism, or epithelioid tumor cells are more easily observed in recurrent or metastatic tumors. Subtype: Hyalinizing spindle cell tumor with giant rosette formation. Immunohistochemical staining: positive for MUC4, epithelial membrane antigen (EMA), CD34, smooth muscle actin (SMA), Claudin-1; negative for desmin, S-100 protein, cytokeratin (CK), β-catenin. 75% of tumors have the specific chromosomal translocation t(7;16)(q33;p11), forming the FUS-CREB3L2 fusion gene; also seen is the specific chromosomal translocation t(11;16)(q11;p11), forming the FUS-CREB3L1 fusion gene and EWSR1-CREB3L1 fusion gene (Figure 3,Figure 4)。
Chest wall hamartoma of infancy
A benign, non-neoplastic lesion presenting as an expansile rib lesion with nodular cartilage, mesenchymal tissue, and secondary aneurysmal bone cyst-like changes. Also known as infantile hamartoma of the chest wall, chondromesenchymal hamartoma, mesenchymal tumor, chest wall mesenchymal hamartoma, infantile vascular hamartoma, etc. Most cases are seen in newborns, 97% of cases are infants under 1 year old, with a male-to-female ratio of 2:1. Lesions are located in the anterior or lateral ribs, occasionally reported in the nasal passage; 9% are multifocal and bilateral lesions[5], often asymptomatic, but may present with difficult labor, respiratory distress, etc.; the mass may grow rapidly. Imaging features include an expansile mass often with chondroid calcification, involving single or multiple ribs; bone destruction often suggests “malignancy.” Ultrasound shows a mixed cystic-solid homogeneous echo. There are occasional reports of tumor progression or regression, but most occur in infants under 1 year old. Surgical resection of the mass may be chosen if symptoms are significant.
The tumor consists of solid and cystic areas. The cystic area resembles an aneurysmal bone cyst: hemorrhagic cavities, cyst walls composed of spindle stromal cells with multinucleated giant cells; cells show no nuclear atypia or abnormal mitotic figures. The peripheral solid part contains hypercellular cartilage nodules with ossification; cartilage is mature, with hypercellular and more primitive morphology. Also seen are chondroblastoma-like proliferation, woven bone, and osteoid tissue areas. Molecular genetic features: one case in a child with Wiedemann-Beckwith syndrome suggests possible involvement of the 11p15.5 ring gene (Figure 5,Figure 6,Figure 7)。
Hyperinsulinism with B-cell ATP-sensitive potassium channel abnormalities
Previously called nesidioblastomatosis. ATP-sensitive potassium channel abnormalities are one of the most common causes of congenital hyperinsulinism, ineffective to channel antagonist diazoxide treatment, requiring surgical pancreatic resection for effective treatment.
Manifests as single or clusters of islet cell hyperplasia with recurrent, persistent, symptomatic hyperinsulinemic hypoglycemia, which can be focal or diffuse, nesidioblastosis. Incidence is 1/50,000 to 1/2,500 newborns; most cases are neonates or infants under 1 year old. Clinical presentation includes recurrent, persistent, symptomatic hyperinsulinemic hypoglycemia, macrosomia, hepatomegaly, polycythemia. Imaging may show normal or slightly enlarged pancreas. The disease is divided into 3 types. Microscopic examination shows pancreatic β-cell hyperplasia and hypertrophy. Diffuse type: pancreas shows enlarged and hyperchromatic β-cell nuclei diffusely distributed, ductulo-insular complexes, islets of various sizes and shapes within pancreatic acinar tissue. Focal type: abnormal islet aggregation, focal or multifocal nodular lesions, large β-cells arranged in bundles, nuclei enlarged and hyperchromatic, β-cells separated by acinar cells; lesions may contain other pancreatic endocrine cells; ductulo-insular complexes are common; adequate sampling is required. Molecular genetic features: mutations in ABCC8 and KCNJ1 genes (β potassium channel proteins); focal type often associated with ABCC8 mutation (Figure 8,Figure 9). The third type is dominant ATP-sensitive potassium channel mutation, rare, with mild clinical symptoms, dominant expression of ABCC8 and KCNJ11 genes.
Fetal lung interstitial tumor (FLIT)
FLIT is a recently recognized solitary, congenital benign space-occupying lesion of the lung, composed of immature airways and stroma, lined by flat or cuboidal epithelium, with underlying bland immature mesenchymal cells. This disease is extremely rare, with less than 15 cases reported in the literature to date. Clinically may be asymptomatic, only discovered by routine prenatal ultrasound; or the infant may present with respiratory distress, feeding difficulties, etc., with or without fever. Most have no specific positive laboratory findings; may have decreased oxygen partial pressure due to hypoxia. Imaging shows a solitary, well-defined mass confined to one lung lobe, solid or cystic-solid (Figure 10). Microscopic examination shows the tumor is well-demarcated from surrounding normal lung tissue, with a fibrous capsule. The tumor consists of variably sized alveolar-like cystic spaces, some of which merge with tubular structures (resembling terminal respiratory bronchioles and alveolar ducts). Cyst walls are lined by flat or cuboidal epithelial cells with pale cytoplasm, bland nuclei without significant atypia. Septa between cysts are widened; stromal cells are oval, polygonal, or spindle-shaped, with abundant clear cytoplasm, round or oval nuclei, fine chromatin (Figure 11). The surface lining epithelial cells are positive for CK, EMA, thyroid transcription factor (TTF)1, and β-catenin. Stromal cells are positive for vimentin, SMA, β-catenin (cytoplasmic staining), Ki-67 labeling index 5%-10%. Desmin, CD34, synaptophysin, HMB45, anaplastic lymphoma kinase (ALK), S-100 protein, CD56, CD30, etc. are negative. FISH detection reveals ALK gene rearrangement in some FLIT cases.
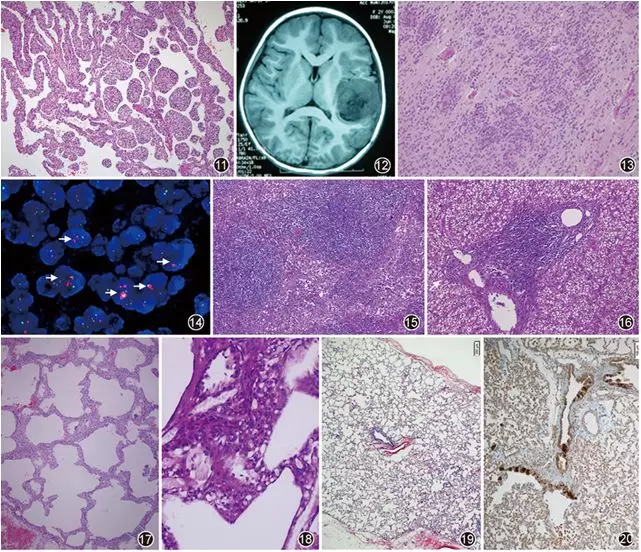
▲Figure 11The mass is composed of immature airways with widened, variably thick septa, slightly spongy. HE, low magnification
▲Figure 12MRI shows a temporal lobe mass.
▲Figure 13Multiple true rosettes. HE, medium magnification
▲Figure 14FISH detection of C19MC gene status (amplification probe). Red dots represent C19MC gene, green dots are reference gene ZNF443. Arrows indicate cells with C19MC gene amplification (C19MC/ZNF443 > 2), showing clustered distribution. FISH, ×1,000
▲Figure 15Shows expansion of the paracortical area of lymph node, lymphoid follicle hyperplasia. HE, low magnification
▲Figure 16Shows lymphocyte infiltration in the portal area. HE, low magnification
▲Figure 17Diffuse thickening of pulmonary interstitium, with slight histiocyte hyperplasia and infiltration in the pulmonary interstitium. HE, low magnification
▲Figure 18Glycogen visible within pulmonary interstitial cells. PAS stain, medium magnification
▲Figure 19Shows normal lobular alveoli and distal small airways. HE, low magnification
▲Figure 20Neuroendocrine cell hyperplasia in distal small airways. Bombesin stain, medium magnification
Embryonal tumor with multilayered rosettes, C19MC-altered (ETMR)
An aggressively growing central nervous system embryonal tumor with multilayered rosettes and alterations (amplification or fusion) of the C19MC gene at 19q13.32. Previously, most childhood central nervous system embryonal tumors were grouped together, including embryonal tumor with abundant neuropil and true rosettes, ependymoblastoma, and medulloepithelioma. Mostly occurs in children under 4 years old. Clinical presentation includes increased intracranial pressure, such as headache, nausea, vomiting, and visual disturbances. CT and MRI show a large contrast-enhancing mass, which may have cystic changes or calcification.
Pathological feature is rosettes, appearing as multilayered, mitotically active pseudostratified neuroepithelium surrounding a central round or slit-like lumen. The lumen formed by rosettes is often empty or contains eosinophilic debris. Some rosettes have a clear inner limiting membrane on the luminal surface. Nuclei of cells forming the rosettes are pushed from the luminal side toward the outer cell border. Most tumors lack an outer limiting membrane of the rosettes. Tumor cells express nestin and vimentin; small cells may focally express epithelial cytokeratin, EMA, and CD99. Usually do not express neuronal and glial markers, but some embryonal tumor cells may express glial fibrillary acidic protein (GFAP). Neuropil and tumor neurons are positive for synaptophysin; Neu N can be positive in tumor neurons. Tumor cells express INI1; Ki-67 labeling index is 20%-80%. LIN28A is a diagnostic marker for ETMR. Molecular genetic characteristics: focal high-level amplification at 19q13.42 is a specific and sensitive marker for this tumor (Figure 12,Figure 13,Figure 14)。
Autoimmune lymphoproliferative syndrome (ALPS)
A disorder of lymphocyte homeostasis due to defects in FAS gene-mediated apoptosis. Clinically classified into: Type I accounts for about 65%, further divided into three subtypes: Ia, germline FAS (TNFRSF6, CD95, APO1) gene mutation; Ib, FAS ligand (FASLG) gene mutation; Is, somatic FAS gene mutation. Type II: mutation in caspase gene. Type III: no gene mutation found in FAS pathway. Type IV: rare, NRAS gain-of-function mutation, patients have ALPS phenotype but FAS-mediated apoptosis is mostly normal. Children often present with chronic non-malignant lymphoproliferation within the first year of life, recurrent or chronic lymphadenopathy; 85% have splenomegaly and hypersplenism, 45% have hepatomegaly, lymphocytic interstitial pneumonia; 70% have autoimmune diseases. ALPS children have increased risk of various malignancies: Hodgkin and non-Hodgkin lymphoma, carcinoma. Non-malignant lymphoproliferative disease in ALPS may regress or improve, with a lifelong risk of lymphoma.
Pathological examination: lymph nodes often show marked expansion of the paracortical area, germinal center hyperplasia (follicular hyperplasia, atrophy), progressive transformation of germinal centers, changes resembling Rosai-Dorfman disease, etc. (Figure 15,Figure 16). Spleen may show expansion of white pulp and red pulp. Liver may show inflammatory lesions in portal areas.
Diagnostic criteria for ALPS: Major criteria: (1) Chronic non-malignant lymphoproliferation; (2) Lymphadenopathy in at least two groups and/or splenomegaly lasting over 6 months; (3) Significant elevation of double-negative T cells in peripheral blood (>5%); (4) Defective FAS-mediated apoptosis in vitro; (5) Mutation in FAS, FASL, CASP10, or NRAS genes. Minor criteria: (1) Autoimmune cytopenia; (2) Moderate elevation of double-negative T cells in peripheral blood; (3) Elevated serum IgG; (4) Elevated serum IL-10; (5) Elevated vitamin B12; (6) Elevated plasma FAS ligand level. Diagnostic standard: three major criteria or two major criteria plus two minor criteria can diagnose ALPS.
Molecular genetic features: mutations in FAS, FASLG, CASP10, etc. have been found in children.
Pulmonary interstitial glycogenosis (PIG)
Mostly occurs in newborns within hours after birth, presenting with sudden onset of tachypnea, dyspnea, and hypoxemia. Chest X-ray shows hyperinflation with diffuse interstitial infiltrates. High-resolution CT: diffuse type mainly shows ground-glass opacities, thickened interlobular septa, and reticular changes.
Microscopic examination: Pulmonary interstitium generally shows no or minimal histiocyte hyperplasia and infiltration; pulmonary interstitium shows diffuse thickening, with abundant vimentin-positive spindle undifferentiated mesenchymal cells, PAS positive. Interstitium is not accompanied by collagen deposition; no inflammatory exudate in alveolar spaces. Spindle undifferentiated mesenchymal cells are PAS positive (Figure 17,Figure 18)。
Neuroendocrine cell hyperplasia of infancy (NEHI)
An interstitial lung disease unique to infancy and early childhood, clinically presenting with persistent dyspnea, pathologically characterized by hyperplasia of neuroendocrine cells in distal airways, often accompanied by formation of neuroepithelial bodies. Commonly seen in healthy full-term or near-term infants (about 85% of patients are full-term), premature infants often have no history of chronic lung disease.
Onset mostly within 1 year of age; generally normal at birth, onset relatively insidious, often gradually developing persistent dyspnea after birth or following infection, lasting from months to years, average 2-3 years, with hypoxemia. High-resolution CT shows characteristic changes: one is geographic distribution of ground-glass opacities, often in right middle lobe and left lingula; another main feature is mosaic pattern of air trapping on expiratory images, often in lower lobes.
Microscopic examination: Lung is normal or shows only mild nonspecific interstitial changes, including airway smooth muscle cell hyperplasia, increase in alveolar macrophages and airway clearance cells. Typical pathological change is increased bombesin-positive pulmonary neuroendocrine cells, mainly distributed in distal small airways, and may cluster within alveolar ducts of lobular parenchyma, forming increased and/or enlarged neuroepithelial bodies. Bombesin and serotonin stains show neuroendocrine cell hyperplasia. Molecular genetic features are related to TTF1/NKX2 gene mutation (Figure 19,Figure 20)。
Acknowledgments
Figures 17-20 provided by Professor Liang Wang, Department of Pathology, Children’s Hospital Los Angeles.
Source:Chinese Journal of Pathology, 2018, 47(11): 817-821. Author: He Lejian