In the previous four articles, we have detailed renal tumors with clear cell morphology, renal tumors with eosinophilic/oncocytic features, and papillary RCC (formerly type 1) and papillary adenoma among tumors with papillary formation. In this issue, we will continue to introduce the remaining subtypes of tumors with papillary formation, as well as renal medullary carcinoma, collecting duct carcinoma, and other aggressive/invasive tumors. We will also provide a brief summary of this series of articles, hoping to serve as a reference for the daily work of our pathology colleagues!

Other Subtypes of Tumors with Papillary Formation
Mucinous Tubular andSpindle Cell Carcinoma
Mucinous Tubular and Spindle Cell Carcinoma (MTSCC) shows significant morphological and immunohistochemical overlap with papillary RCC. AMACR positivity is common in both, while CK7 staining is variable in MTSCC. The triad of tubular growth, spindle cell morphology, and mucinous stroma often aids in diagnosing MTSCC on histological grounds. Cytogenetic analysis reveals that MTSCC frequently exhibits chromosomal losses, whereas type 1 papillary renal cell carcinoma often shows gains of multiple chromosomes, specifically chromosomes 7 and 17. Overexpression of VSTM2A and IRX5, assessed by RNA in situ hybridization, has recently been found to be highly specific for MTSCC, but it is not yet widely used.
Inverted Papillary Renal Tumor
Recently, it has been recognized that a subgroup of tumors with recurring distinct immunohistochemical and molecular features may need to be classified as a distinct tumor type. This tumor has eosinophilic cells, exhibits papillary and tubular growth, and features nuclei located above the base, termed inverted papillary renal tumor. This tumor consistently shows CK7 positivity. Compared to typical papillary renal cell carcinoma, AMACR is variable and usually less diffuse. GATA3 is positive, and vimentin is negative. It is important to note that GATA3 can be expressed in clear cell papillary RCC, chromophobe RCC, and other rare renal tumors. Next-generation sequencing reveals recurrent KRAS gene mutations, which are currently not considered present in any other recognized renal tumor, indicating this is a distinct entity among renal tumors.
Papillary Renal Cell Carcinoma (formerly type 2), FH-deficient Renal Cell Carcinoma, and High-grade Papillary Tumors
Traditionally, type 2 papillary renal cellcarcinoma was considered a separate entity, but there is increasing recognition that it represents apatternencountered in multiple distinct entities.Tumors with high-grade nuclei and eosinophilic cytoplasm, featuring multilayered and stratified tumor cells, can be seen in various entities, including but not limited to TFE3-RCC, FH-RCC, and TFEB-amplified RCC.FH-RCC is an aggressive renal tumor with potential genetic significance because it is overwhelmingly seen in HLRCC.This diagnosis is suggested when encountering a large unilateral infiltrative mass, predominantly papillary but also with variable solid, tubular, and cystic components.Tumor cells typically have eosinophilic cytoplasm and nuclei with prominent nucleoli, sometimes with perinuclear clearing (Figure 1A).Although more than half of the tumors primarily exhibit papillary features, cases with significant morphological similarity to tubulocystic renal cell carcinoma have also been reported.
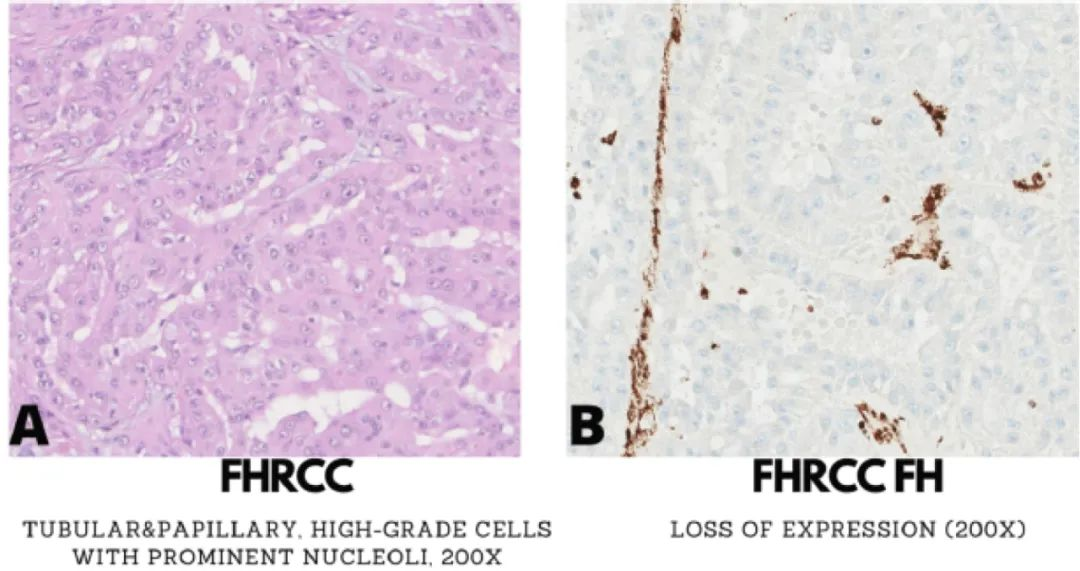
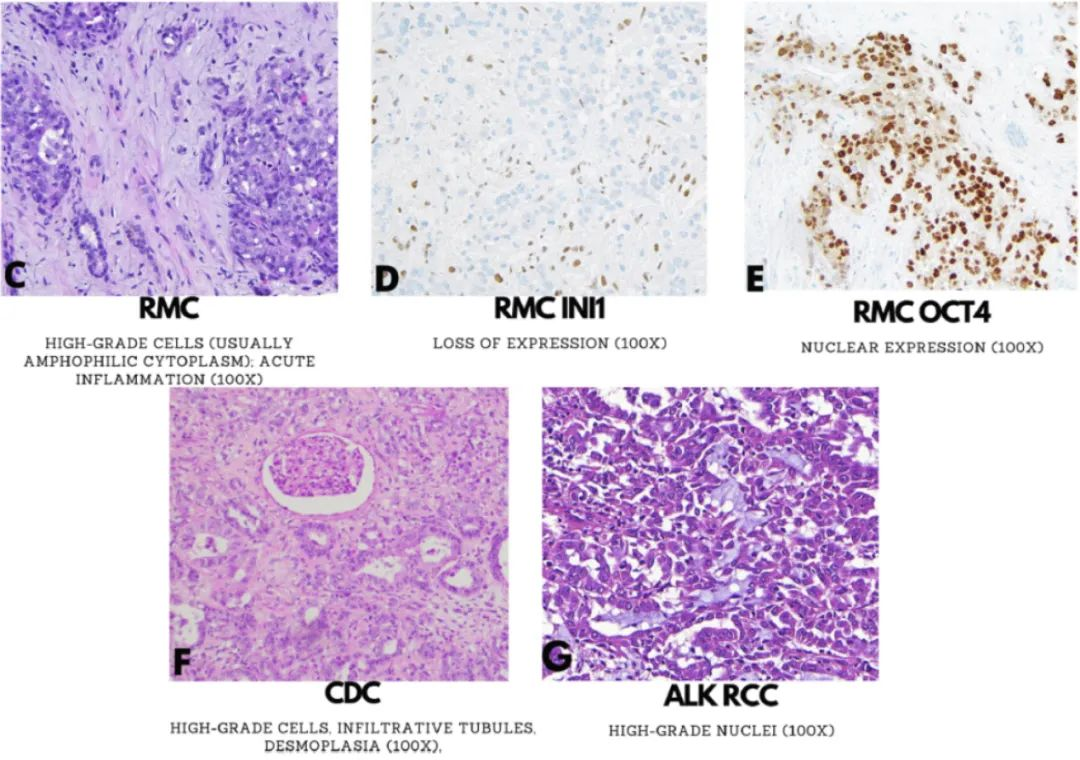
Figure 1. Morphological and immunophenotypic features of fumarate hydratase-deficient papillary renal cell carcinoma (FH-RCC; A-B), renal medullary carcinoma (RMC; C-E), collecting duct carcinoma (CDC; F), and anaplastic lymphoma kinase gene rearrangement-associated renal cell carcinoma (ALK-RCC; G).
FH-RCC is negative for CK7 and p63, but approximately half of the cases can express GATA3, posing a potential risk for misdiagnosis as high-grade urothelial carcinoma. FH negativity (Figure 1B) and/or 2SC positivity are considered supportive of FH-RCC. TFEB-amplified RCC is another aggressive tumor; TFEB expression is similar to that in TFEB gene rearrangement counterparts. TFEB-amplified RCC has a heterogeneous IHC profile, often positive for Melan A and cathepsin K, and negative for CA IX compared to clear cell RCC.
Renal Medullary Carcinoma, Collecting Duct Carcinoma, and Other Aggressive/Invasive Tumors
Rare, medulla-centered high-grade aggressive tumors thought to recapitulate the distal nephron include renal medullary carcinoma (RMC) and collecting duct carcinoma (CDC), with few other primary renal origins in the differential diagnosis (Table 1). PAX8 is consistently positive in these renal tumors, although it is always important to consider urothelial carcinoma or metastases from other cancers before making these diagnoses. In particular, urothelial carcinoma may be PAX8 positive, so other urothelial markers such as GATA3 and p63 should typically be used in combination. Hemoglobinopathy, usually sickle cell trait, is a prerequisite for diagnosing RMC. Recently, rare morphologically identical tumors without hemoglobinopathy have been identified, leading to the proposed term ‘unclassified RCC with medullary phenotype.’ Infiltrative, often fused tubules seamlessly infiltrate benign renal parenchyma with high-grade tumor cells, often accompanied by intraluminal acute inflammation (Figure 1C). Alterations including frequent INI-1 gene translocations are found in RMC, leading to aberrant loss of INI-1 protein in almost all tumors (Figure 1D). OCT4 (also known as OCT3/4) is also positive in many RMCs (Figure 1E). CK7 and CK20 may be co-expressed, so the assumption that this expression profile is specific to urothelial carcinoma is not robust.
Table 1. High-grade Invasive Renal Tumors
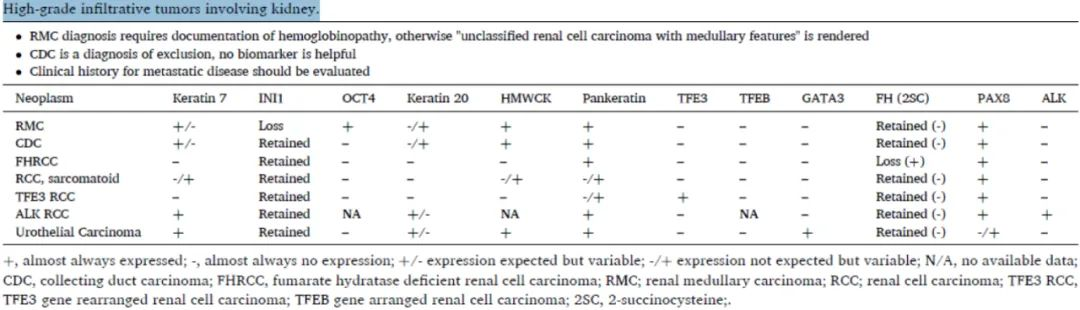
(Click to view larger image)
Once RMC (or unclassified RCC with medullary phenotype), urothelial carcinoma, FH-RCC, or secondary organ metastases are excluded, any other primary renal carcinoma should be considered a diagnosis of exclusion. For example, CDC lacks reliable markers and can only be diagnosed based on some non-specific histological features, such as predominant tubular formation, desmoplasia, infiltrative growth, and exclusion of other primary and secondary entities (Figure 1F). Some authors have reported p63 positivity in CDC; however, this usually favors urothelial carcinoma. FH-RCC predominantly shows infiltrative renal tubules and can be distinguished by FH-/2SC+. Another tumor in this category, currently provisional, is ALK-RCC, which has heterogeneous morphological features, often with rhabdoid morphology and a mucinous background (Figure 1G). Rare metanephric adenomas closely resemble ALK rearrangements, but their significance is unclear. ALK-RCC can usually be detected by ALK IHC, which has important therapeutic implications given available targeted therapies. If specific features of any of the above entities are not identified, the remaining primary renal cell carcinomas with invasive features may be diagnosed as unclassified renal cell carcinoma.
In many cases, the classification of renal tumors can be based solely on histological morphology. However, for tumors that raise two or more differential diagnoses, a small panel of immunohistochemical markers can often narrow down or resolve the differential diagnosis, aiding in most diagnostically challenging pathology specimens. For confirming renal origin, PAX8 is the most reliable marker (although it is important to be aware of some limitations). Other useful markers include CA IX (clear cell type), AMACR (papillary), KIT (chromophobe RCC and oncocytoma), CK7 (papillary, clear cell papillary, and chromophobe RCC), and vimentin (non-reactive in oncocytoma/chromophobe carcinoma). Abnormal loss of staining for SDHB, FH, and INI-1 helps in diagnosing SDHB-deficient, FH-deficient, and medullary carcinoma, respectively.
Reference: Akgul M, Williamson SR. Immunohistochemistry for the diagnosis of renal epithelial neoplasms. Semin Diagn Pathol, 2022 Jan;39(1):1-16.
